





Inventamos para la vida
Seguimos la ciencia para hacer frente a algunas de las mayores amenazas para la salud en todo el mundo. Te invitamos a conocer qué dicen nuestra gente y nuestros pacientes sobre nosotros.
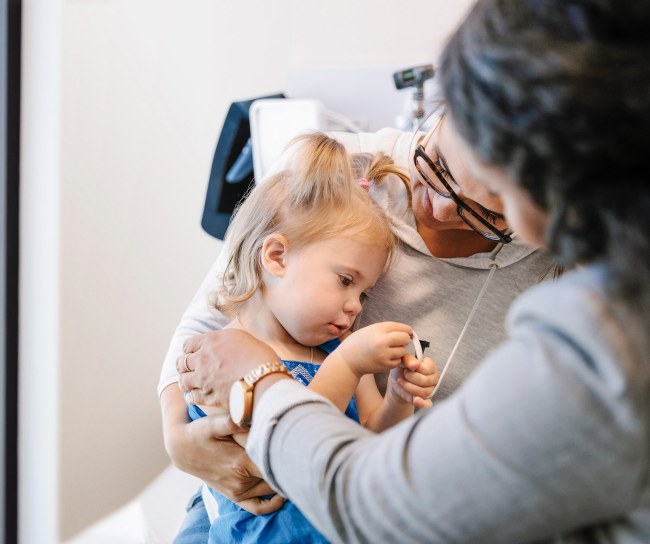
Tu bienestar es nuestro propósito
Trabajamos duro para brindarte los medicamentos y vacunas que ayudan a combatir algunos de los problemas de salud más graves que enfrentamos.

Donde la creatividad se reúne con la investigación
Reunimos mentes creativas para inventar medicamentos y vacunas importantes. Nada nos detiene para ayudar a salvar y mejorar vidas.

Oncología
Nuestra misión es brindar innovaciones que extiendan y mejoren la vida de las personas con cáncer.

Vacunas
Las vacunas son una de las mayores historias de éxito en la salud pública, y hemos descubierto, desarrollado y lanzado vacunas que ayudan a prevenir enfermedades por más de 100 años.

Enfermedades infecciosas
Desempeñamos un papel importante en el descubrimiento y desarrollo de medicamentos y vacunas innovadores para tratar y prevenir enfermedades infecciosas, como el VIH y el ébola.

Trastornos cardiometabólicos
Estamos decididos a encontrar soluciones para los desafíos crónicos de salud más graves, como las enfermedades cardiovasculares y la diabetes.

Descubrimiento y desarrollo
Seguimos la ciencia donde podemos hacer la mayor diferencia, ahora y en el futuro.


Responsabilidad corporativa
Estamos apoyando el futuro de nuestro negocio y el bienestar de los pacientes, las personas y las comunidades de todo el mundo.